Analyser les génomes bactériens : Du séquençage à la génomique comparative
L’avènement des technologies de séquençage à haut débit (NGS) a transformé la microbiologie, rendant l’analyse d’un génome bactérien complet accessible en routine. L'analyse ne se limite plus à la simple lecture des bases (A, T, G, C) : elle repose sur un pipeline bioinformatique rigoureux, allant du contrôle qualité à l’interprétation évolutive. Ce guide vous présente les étapes incontournables pour décrypter et valoriser un génome bactérien.
🔬 1. Séquençage : choisir la bonne technologie
Le séquençage produit des millions de lectures (reads) à partir d’ADN génomique extrait. Deux grandes familles de technologies coexistent :
- Lectures courtes (short‑reads) : Illumina (HiSeq, NovaSeq) offre une très haute précision (taux d’erreur < 0,1 %), avec des reads de 150–300 pb. Idéal pour le séquençage d’isolats et la détection de variants.
- Lectures longues (long‑reads) : Oxford Nanopore (MinION, PromethION) et PacBio (SMRT) génèrent des lectures de 10–100 kb. Elles sont indispensables pour résoudre les régions répétées (opérons d’ARNr, transposons) et obtenir des génomes circulaires complets (« gapless »).
L’approche hybride (Illumina + long‑reads) combine précision et contiguïté, idéale pour des génomes de référence de haute qualité.
🧩 2. Assemblage de novo : reconstruire le génome
L’assemblage consiste à fusionner les lectures pour recréer les chromosomes et plasmides. Selon le type de données :
- Assembleurs pour short‑reads : SPAdes (très populaire pour les bactéries), ABySS, MEGAHIT utilisent des graphes de de Bruijn.
- Assembleurs pour long‑reads : Flye, Canu, hifiasm (pour PacBio HiFi) exploitent le chevauchement lecture‑lecture.
- Assembleurs hybrides : Unicycler (spécialisé pour les génomes bactériens) combine Illumina et Nanopore/PacBio pour produire des contigs circulaires.
Après assemblage, la circularisation manuelle ou automatique (ex. circulator) permet de fermer le chromosome. La qualité est évaluée avec QUAST : N50, nombre de contigs, longueur totale.
🏷️ 3. Annotation structurale et fonctionnelle
L’annotation identifie les éléments génomiques : gènes codant pour les protéines (CDS), ARNt, ARNr, et attribue une fonction biologique. Les pipelines automatisés les plus utilisés :
- Prokka : rapide, conçu pour les génomes bactériens et archées ; intègre Prodigal pour la prédiction des CDS et des bases de données comme UniProt, RefSeq.
- RAST (ou RASTtk) : interface web complète, apprécié pour les débutants.
- Bakta : annotateur moderne, très complet (plasmides, CRISPR, gènes de résistance).
L’annotation fonctionnelle repose souvent sur l’alignement via BLAST contre des bases de référence (COG, KEGG, EggNOG), et l’assignation de termes GO pour les analyses d’enrichissement.
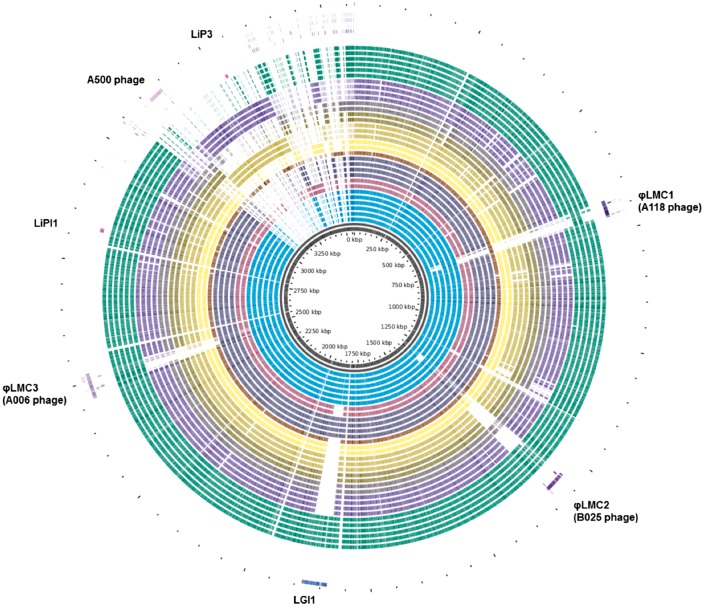
🧬 4. Génomique comparative et pan‑génome
Comparer plusieurs souches d’une même espèce permet de comprendre la plasticité génomique. On distingue :
- Core‑genome : ensemble des gènes présents chez toutes les souches (généralement lié au métabolisme central).
- Pan‑génome : totalité des gènes distincts rencontrés dans l’espèce (core + accessoire + gènes uniques).
- Génome accessoire : gènes présents dans certaines souches seulement, souvent acquis par transfert horizontal (plasmides, prophages, îlots génomiques).
Outils de référence : Roary (rapide, pan‑génome), Panaroo (robuste aux erreurs d’annotation), BPGA (analyse fonctionnelle). L’inférence phylogénétique peut se faire à partir du core‑génome (SNP) ou de matrices de présence/absence.
🔍 Recherche de gènes spécifiques : résistance et virulence
Des bases de données dédiées comme CARD (résistance aux antibiotiques) ou VFDB (facteurs de virulence) sont interrogées via ABRicate ou ResFinder. L’identification des îlots génomiques est possible avec IslandViewer ou GIPSy.
📊 5. Visualisation et interprétation finale
La navigation dans le génome annoté est facilitée par des interfaces graphiques :
- Artemis : affichage détaillé des annotations, manuel ou automatique.
- JBrowse : visualisation web interactive, idéale pour le partage de données.
- BRIG (BLAST Ring Image Generator) : représentation circulaire comparative de plusieurs génomes autour d’un génome de référence.
L’analyse finale intègre les résultats d’assemblage, d’annotation, de résistance et de phylogénie pour proposer une description complète de la souche étudiée (ex. publication d’un génome de référence).
💬 Commentaires